Osteogenesis imperfecta treatment regimens in children. Metabolic - hereditary metabolic diseases
Osteogenesis imperfecta (Lobstein-Frolik disease, congenital bone fragility, periosteal dystrophy) is a group of genetic pathologies characterized by impaired bone tissue formation. Then the child’s bone fragility increases, and as a result, pathological fractures occur. In addition, bones become deformed, muscles become thinner, joint hypermobility occurs, hearing is impaired, etc.
The congenital form of the disease is the most dangerous; it has a severe course and leads to death from numerous complications. The prognosis for the late form is more favorable. It is impossible to completely cure the pathology. Supportive treatment is carried out to help strengthen bone tissue and prevent fractures.
Description of the pathology
Lobstein-Frolik disease is a genetically determined disease that occurs as a result of impaired bone formation. It leads to a decrease in bone mass and increased fragility. The pathology develops as a result of a defect in type 1 collagen, which is an important protein in the bone structure. Then it is produced in insufficient quantities or the structure of the substance is disturbed. For this reason, bones become weak and brittle. Because of this, the pathology is called “crystal disease.”
Reference. According to statistics, in approximately 50% of cases, imperfect bone formation is provoked by spontaneous mutations. The disease is diagnosed in 1 child out of 10–20 thousand newborns.
Crystal disease is incurable, but with the right approach it can make a child’s life much easier.
Symptoms
Symptoms depend on the type of pathology.
Osteogenesis imperfecta is manifested by pathological fractures, bone deformation
The early form of the disease is the most dangerous, as sometimes children die in the womb. Most newborns die in the first days or months of life. This is associated with intracranial birth injuries, severe respiratory disorders, and acute respiratory viral infections.
Osteogenesis imperfecta in children is manifested by the following symptoms:
- Thin, pale skin, thinning subcutaneous fat.
- General weakness, hypotension.
- Bone fractures (femur, lower leg, forearm, shoulders) with minimal impact.
Usually, in the early form of the pathology, the child dies within 2 years.
The late form is manifested by the following symptoms:
- Increased bone fragility.
- Blue discoloration of the whites of the eyes.
- Hearing impairment, up to complete deafness.
- Late overgrowth of the fontanel.
- Slowing down of the child's physical development.
- Excessive joint flexibility due to weak ligaments.
- Muscle thinning.
- Dislocations, fractures with minimal impact.
- Curvature or shortening of bones after their fusion.
- Deformation of the sternum or spine.
- Late teething (after 1.5 years), dental anomalies, caries, rapid abrasion and destruction of teeth, staining them yellow.
- Hearing impairment, deafness.
Crystalline disease may be accompanied by bulging of the wall of the mitral valve of the heart or its functional failure, kidney stones, inguinal hernias, nasal hemorrhages, etc.
Classification of crystalline disease
There are 2 known forms of pathology:
- Congenital. Fractures occur in the womb and immediately after birth.
- Late. Bones are injured when the child is already walking. This form of the disease has a milder course.
Types of crystal disease:
- Osteogenesis imperfecta type 1 - fractures occur after birth until adolescence, the spine is slightly curved, the ligaments and joints are weak, and muscle tone is reduced. The whites of the eyes become discolored, children lose their hearing early, and their eyes are slightly bulging.
- Type 2 – the development of the skeleton is disrupted, the bones are deformed or shortened, and protrusions remain at the sites of fractures after fusion of the bone tissue. Children develop slowly physically. This type of disease is considered the most severe. A child may die before the age of 1 year from functional lung failure or hemorrhage into the cranial cavity. The bones are severely deformed, the patient is short in stature.
- Type 3 – bones are injured after birth until puberty. Severe deformations of the bones, spine, chest, breathing problems, weak muscles, joints and ligaments are possible. The sclera becomes discolored, and hearing impairment quickly progresses.
- Type 4 – symptoms of bone development disorders are practically not noticeable, but patients develop premature osteoporosis (decreased bone density). Fractures are typical before adolescence, the curvature of the bones is of mild or moderate severity. The patient is short in stature and may lose hearing early.
- Type 5 – the course of the disease is the same as in type 4 pathology. The only difference is that the bone has a mesh structure.
- Type 6 – symptoms are the same as for type 4 disease, but the bone structure resembles fish scales.
- Type 7 – disorders associated with mutations in cartilage tissue.
- Type 8 – there is a strong change in the protein, which contains leucine and proline (amino acids). This type of pathology has a severe course and ends in death.
Reference. Depending on the type of inheritance, autosomal dominant and autosomal recessive osteogenesis imperfecta are distinguished. The first type is typical for types 1 – 5 of pathology, and the second – for types 7 – 8.
Causes of Lobstein-Frolik disease
The causes of osteogenesis imperfecta are associated with genetic pathologies. The gene for collagen A1 and A2 mutates, causing a lack of protein or its structure being disrupted. Then the fragility of bone tissue increases, especially the tubular bones (shoulders, forearms, thighs, legs) suffer. They have a porous structure, bone islands, a large number of sinuses, which are filled with loose tissue, the outer layer is thinned.
Doctors distinguish 2 types of inheritance of crystalline disease:
- Autosomal dominant - the disease is passed on to the child from one parent who also suffers from it. Then bones are more often injured after 1 year.
- Autosomal recessive - a mutated gene is passed on from both parents. The disease has a severe course, pathological fractures are possible in the womb or immediately after birth.
Reference. Osteogenesis imperfecta with an autosomal dominant type of inheritance is more often diagnosed.
Establishing a diagnosis
The congenital form of the pathology can be detected as early as 16 weeks of pregnancy using ultrasound. If necessary, a chorionic villus biopsy and gene diagnosis are performed to confirm the presence of a mutated gene.
In other cases, the diagnosis of osteogenesis imperfecta consists of the following methods:
- Collection of anamnesis, patient complaints. Signs of pathology: frequent fractures, abnormal bone shape, difficulty gait, short stature, bad teeth, hearing impairment.
- Visual inspection. The doctor evaluates height, body weight, hearing, condition of teeth, color of the whites of the eyes, and conducts neurological tests. The orthopedist is interested in the shape, length of the limbs, deformations, range of motion in the joints.
- Laboratory tests of blood and urine will help detect the level of proteins, glucose, urea, calcium, phosphorus, etc.
- An X-ray of the limbs, spine, skull will show that bone density has decreased, bone calluses after healing of pathological fractures, etc.
- A bone biopsy (examination of a fragment of bone tissue) is used to confirm a decrease in its density and thinning of the outer layer.
- A skin biopsy is performed to examine the collagen defect.
- Molecular genetic testing will help detect the mutated gene. To do this, the patient's blood or saliva is studied.
Reference. Differential diagnosis will help to distinguish crystalline disease from rickets (a malformation of the cartilage-forming system of the fetus), desmogenesis imperfecta (hyperelasticity of the skin).
Treatment methods
As already mentioned, osteogenesis imperfecta is incurable. Treatment is carried out to alleviate the patient’s condition and strengthen bone tissue. For this purpose, the following methods are used:
- Drug therapy. The patient takes drugs based on somatotropin (growth hormone) to stimulate collagen synthesis. In addition, antioxidants, medicines containing calcium, phosphorus, and vitamin D2 are indicated.
- Then the patient is prescribed drugs that accelerate the formation and mineralization of bone tissue, which contain extract of the thyroid glands of cattle and cholecalciferol. And bisphosphonates slow down the process of bone destruction; pamidronic acid, zoledronic acid, and residronate are used for this purpose.
- Physiotherapeutic procedures: electrophoresis with calcium chloride (penetration of a medicinal substance through the skin using an electric current), ultraviolet irradiation of blood, magnetic therapy, inductothermy, etc. Children are also prescribed massage, therapeutic exercises to strengthen muscles and ligaments.

Medicines will help strengthen bone tissue and alleviate the patient’s condition
In addition, the patient may need treatment from a psychologist. The use of orthopedic devices, such as shoes or corsets, is also recommended.
In case of severe bone deformation after fractures, a corrective osteotomy is performed. The surgery helps correct the shape and size of the limbs. During the procedure, the affected bone is dissected, the irregular shape is corrected, and bone fragments are fixed with special pins or bolts (osteosynthesis).
There are 2 types of osteosynthesis: bone and intramedullary. In the first case, the fixation structure is located in the patient's body, but outside the bone. The disadvantage of this treatment method is that the periosteum is damaged. In the second case, the fixator is placed inside the bone.
Attention. Surgery for osteogenesis imperfecta is contraindicated if the patient’s condition is severe, he suffers from functional failure of the heart, lungs, or it is impossible to fix the fixator due to lack of bone tissue.
The most important thing
Thus, the most dangerous form of pathology is considered to be the early one, in which most children die within the first months or years. This occurs due to multiple injuries and infections (pneumonia, sepsis). The late form of crystalline disease has a more favorable prognosis, although the quality of life is reduced. Maintenance drug therapy will help get rid of the symptoms of the pathology, strengthen bone tissue, and improve the general condition of the patient. In case of severe bone deformation due to fractures, a corrective osteotomy is performed. Treatment is complemented by physiotherapy, exercise therapy, and massage. Doctors strongly recommend medical genetic counseling for expectant mothers whose families have patients with osteogenesis imperfecta.
3021 0
Diseases of bones and joints can affect even medical professionals who have extensive experience in their field and have achieved a certain level of skill.
But even experienced doctors are horrified when they see a newborn “crystal” person.
Concept and statistics
Osteogenesis imperfecta or “crystal man disease” is a serious disorder of the intrauterine development of bones and joints.
The disease is characterized by increased fragility of bones and joints of the human skeleton. An explanation for the presented disease can be found in the lack of collagen in the patient’s body, or inconsistency with the norm of the described important protein in the structure of the bone skeleton.
The disease is a genetic manifestation and occurs in children whose parents also suffer from the pathology.
In rare cases, the disease is diagnosed in children of completely healthy parents and relatives. Such features are explained by spontaneous mutation.
Statistics of the disease can lead pregnant women into a state of panic, since the rate of fixed cases is 1 newborn child per 15 thousand of all pregnancies.
You should not give in to emotions, since modern medical research and treatment methods can lead to positive results in the recovery of a sick child.
Causes of the disease
As already mentioned above, the described disease is a consequence of a hereditary mutation of collagen genes, which leads to a violation of its structure or lack of it in the body.
Also, the manifestation of “crystal man disease” is affected by a lack of synthesized collagen. 
This type of disease occurs in a milder form and is characterized by an increased degree of bone fragility, which leads to frequent fractures in the sick person. After puberty, the number of fractures decreases significantly, and in adulthood everything repeats.
Experts cannot explain the reasons for the spontaneous mutation. The only thing that can be advised to pregnant women is to be attentive to their health and undergo regular examinations during pregnancy.
Do not drink alcohol and stop smoking while a person is in utero.
Types and symptoms of the disease
Osteogenesis imperfecta has several forms of manifestation and development, which are characterized by distinctive symptoms and the structure of the bones of the sick person.
Type I – weak form
The number of people suffering from this particular type is approximately 50% of all identified cases. As already mentioned, patients are susceptible to frequent bone fractures and joint dislocations.
The risk of fractures decreases after 10 years of life, but after 40 years of age the patient returns to the risk group.
With the first type, certain changes occur in the aorta, as a result of which frequent nosebleeds can be observed.
Type II – perinatal-lethal
This form of manifestation of osteogenesis imperfecta is characterized by frequent fetal death during a woman’s pregnancy. Otherwise, premature birth occurs at short stages of pregnancy. There are also three groups here:
- Group A– head injuries are recorded even at the stage of intrauterine development. Children are born only 20-30 cm tall. Disorders of brain activity and the respiratory system are clearly expressed. Newborns are either stillborn or die within the first few days (in rare cases, dying at the end of the first month of life). The child's death was caused by numerous fractures.
- Group B– signs of the disease are the same as in group A, with the exception of normal development of the respiratory system or with slight deviations from the norm. Such newborns can live for several years. They have shortening of all tubular bones.
- Group B- Diagnosed very rarely. Newborns die in the first days of life or are already born dead. Thinning of the tubular bones and lack of ossification of the skull are noted.
Type III - bone growth disorder
Type III is extremely rare and is characterized by impaired bone growth.
The body of a newborn with short stature can have normal weight. Circulatory disorders are also diagnosed, which in most cases causes death. Bone fractures are recorded during childbirth.
Type IV - skeletal growth disorders
Type IV is characterized by the presence of skeletal abnormalities. After a few years, the patient develops bone calluses, and the number of fractures decreases. After 30, hearing loss is observed.
Comprehensive diagnostics, which is carried out immediately after the birth of the baby, helps to identify the type or group of pathology.
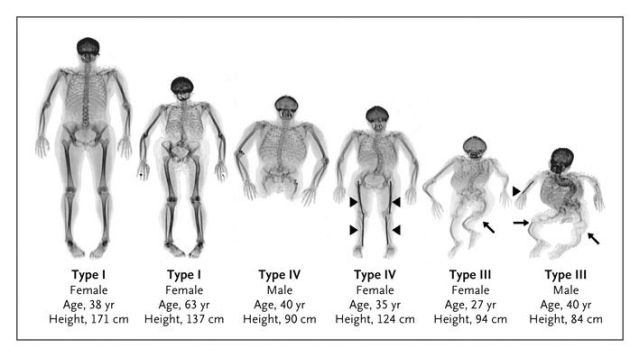
The photo shows four types of osteogenesis imperfecta
Diagnosis of the disease
Diagnosis occurs in two stages. It should also be noted that there is intrauterine diagnosis. Ultrasound is used here.
If there are suspicions of violations of the collagen structure, an additional series of chemical examinations are carried out with the collection of amniotic fluid and epithelial tissue from the pregnant woman.
Immediately after the birth of a sick baby, a number of instrumental studies are carried out, which highlight:
- X-ray – using the image you can identify existing fractures;
- densitometry – the study of the mineral cavity of bone tissue is carried out;
- Bone tissue is collected for biopsy.
In addition to instrumental tests, laboratory tests are also carried out:
- based on blood, abnormalities in the DNA structure are detected;
- carry out tests for collagen diagnostics;
- perform several tests based on a skin biopsy.
Based on the diagnosis, specialists draw up a suitable treatment plan.
Treatment of pathology
 Osteogenesis imperfecta is treated using several methods, including a medicinal method based on the use of drugs by the patient to increase bone density in order to reduce the number of fractures.
Osteogenesis imperfecta is treated using several methods, including a medicinal method based on the use of drugs by the patient to increase bone density in order to reduce the number of fractures.
The composition of medications should include calcium, vitamin D, potassium salts, magnesium and other useful chemicals.
In addition to drug therapy, physiotherapy and therapeutic exercises are used. Also, the treatment is based on psychotherapy, which is carried out by the parents of the sick child and other relatives.
The task of psychotherapy is to explain the basic rules and methods for further teaching the child how to behave in society, so that he can independently avoid situations that could lead to a fracture.
Complications and prognosis
It is impossible to prevent and protect your child from the development of pathology, but with timely treatment and implementation of all the advice and recommendations expressed by the doctor, newborn babies can grow up and be realized in life, since characteristic psychological or mental abnormalities are not detected in children. 
Complications can be caused by a fall, because for a “crystal” person, a small hit on the back with a ball while playing football can result in a spinal fracture.
That is why the life of crystal people cannot be compared with the life of healthy people.
They must protect themselves from the slightest injuries and bruises; any fall can lead to a broken leg and subsequent placement in a wheelchair.
If parents accept their child and begin appropriate actions for his recovery, good results and positive dynamics can be achieved.
As a rule, a complete recovery will not follow, but thanks to modern technological progress, the patient’s life can be significantly made easier.
Congenital bone fragility is a genetically determined disease that affects connective tissue, and, in particular, causes disturbances in the structure and function of collagen.
As the name suggests, it is associated with very poor bone structure, which becomes brittle and brittle.
Osteogenesis imperfecta - causes
Collagen is an important element of the body's connective tissue. Depending on where this connective tissue is located and what functions it performs, the corresponding type of collagen is included in its composition.
Osteogenesis imperfecta is a disease that has a genetic background, and the mutation concerns one gene responsible for the correct structure of collagen - more precisely, responsible for the synthesis of the alpha chain of collagen 1. Disturbances in the structure of this type of collagen impair the structure and strength of bones, tendons, skin, and sclera .
The disease is inherited more often according to an autosomal dominant pattern. This means that the presence of one defective gene from one of the parents is enough to cause symptoms of the disease.
Depending on the extent to which the mutation occurs, the severity of the symptoms associated with the disease changes. The mutation can only slightly reduce the collagen content of connective tissue, which is reflected in the mild course of the disease. Significant disturbances involving the process of collagen synthesis can cause very serious health conditions.
In some people, the disease is inherited in an autosomal recessive manner, meaning that both genes from both parents must be damaged for symptoms to appear. This can happen if each parent is a carrier of a genetic mutation.
Symptoms of congenital bone fragility
Even if the disease is found in several family members, its course may differ significantly. All cases, however, are characterized by varying degrees of weakening of the bone structure, increased bone fragility, and a tendency to fracture. Damage can occur from a bruise, which would not cause anything in a healthy person, and in extremely severe cases, bone damage occurs even at rest.
Other common symptoms include:
- short stature
- bone deformities
- dental hypoplasia
- Blue eyes
- deafness (in adulthood)
- joint laxity
- instability of joints and ligaments
- frequent bruises
Types of congenital bone fragility
There are several types of congenital bone fragility. The mildest form is 1 type, the most common, characterized by low bone fragility, children are not too small and bone deformities are not observed. When a child begins to take his first steps, the first fractures appear, including not only long bones, but also small bones of the arms or legs.
Unfortunately, this tendency accompanies the child until he reaches adulthood. As an adult osteoporotic changes in bones appear much earlier than in healthy people, and hearing loss increases.
Type 2 congenital bone fragility is called lethal form. The disease manifests itself already during intrauterine life, which leads to pronounced fractures and deformities, often ends in fetal death. Having a baby with brittle bone type 2 gives him little chance of surviving for several years. As a rule, such children die at an early age.
Types 3-4– this is a moderate and severe form, when pronounced bone deformations are observed, the disorders have different shapes and severity. They are intermediate forms between the weak form and the lethal form and can give a varied clinical picture.
Bone damage can appear before the baby is born. In more severe cases, the child’s growth is delayed and his posture is deformed. Such people use wheelchairs because their illness makes it impossible to function normally.. Such people develop deafness much earlier. Patients require constant orthopedic consultations.
Treatment of congenital bone fragility
Due to the genetic origin of the disease, there is no possibility of a complete cure and effective therapy.
Treatment focuses on minimizing the number of fractures, preventing major deformities, and reducing pain. Bisphosphonate therapy is used, that is, drugs used in the treatment of osteoporosis. The hearing organs are monitored and the patient’s general condition is assessed to rule out problems in other body systems.
The life expectancy of patients with congenital bone fragility depends primarily on the type of disease. In type 2, the patient may not differ from the general population. In more severe forms, life expectancy is shorter than in the general population, however, this does not result from frequent fractures, but from concomitant problems with the functioning of the respiratory or cardiovascular system, which is associated with chest deformation.
There are several different osteogenesis imperfecta syndromes. The diseases are characterized by damage to the bones, eyes, teeth, ears and cardiovascular system. Their classification is based on the type of inheritance and clinical manifestations.
Osteogenesis imperfecta type 1
This type is the most common. It is inherited in an autosomal dominant manner and is characterized by pronounced intrafamilial variability. The patient may be short, have frequent fractures and a pronounced decrease in working capacity, while his close relative with the same disorder can live a full life. The cause of this syndrome may be defects in both alpha 1(0) and alpha 2(1) procollagen. Mutations often manifest themselves as blue sclera and short stature.
Osteogenesis imperfecta type 2
Type 2 combines the classic congenital variants, in which almost all patients die in childhood or in utero). Many cases are the result of a new mutation (dominant transmissible phenotype if the patient survives and is fertile) in procollagen alpha 1(1) or alpha 2(1). The "dominant negative" model explains the severe phenotype resulting from a heterozygous mutation. Sometimes siblings of patients have the same symptoms with healthy parents. In some cases, mutations were found in the gonads in loci with low levels of expression, which is a risk of having several sick children.
Osteogenesis imperfecta type 3
Type 3 is manifested by severe skeletal deformation, kyphoscoliosis, short stature and frequent fractures of various locations. It usually develops sporadically, which means the appearance of new mutations or an autosomal recessive mode of inheritance.
Osteogenesis imperfecta type 4
Type 4 is phenotypically and genetically similar to type 1, is less common, does not present with blue sclera, and is associated with fewer fractures.
Symptoms
The medical history for fractures is usually similar. “Brittle bones” is a universal manifestation. Sometimes fractures occur in utero, especially in type 2, which makes antenatal radiographic diagnosis possible. In such cases, at birth the limbs are short and curved, multiple rib fractures give a “rosary” appearance on radiographs. Patients with type 1 or 4 usually have a history of few fractures, although blue sclera, opalescent tooth enamel, or hearing loss indicate the presence of the mutant gene. Fragility and deformability are the result of a defect in the collagen matrix of bones. Therefore, skeletal manifestations of osteogenesis imperfecta are a hereditary form of osteoporosis. In elderly patients with age-related or postmenopausal changes, or in young patients with prolonged immobilization after fractures or orthopedic surgeries, fishbones (depressions and ulcerations on the smooth upper and lower edges of the vertebrae due to the pressure of a tensile intervertebral disc) or flat vertebrae are often noted.
The incidence of fractures decreases during puberty in patients with types 1, 3 and 4 disease. Sometimes after a fracture, the formation of false joints occurs. Also quite often, patients develop hypertrophic callus, which is difficult to distinguish from. The issue of an increased risk of osteosarcoma against the background of osteogenesis imperfecta remains controversial, but the risk is still low, but if pain occurs in the absence of a fracture, especially in elderly patients, it is always necessary to exclude osteosarcoma. Relaxation of the ligamentous apparatus of the joints is most pronounced in type 1 osteogenesis imperfecta. Dislocations are the result of deformation due to repeated fractures, relaxation of ligaments or rupture of tendons.
Diagnostics
Differential diagnosis of osteogenesis imperfecta includes idiopathic juvenile osteoporosis, Hajdu-Chinei syndrome (osteoporosis, multiple intercalary bones of the skull, acroosteolysis), pycnodysostosis (dwarf stature, brittle bones, absent mandibular rami, cleft fontanelles, acroosteolysis), and hypophosphatasia. In one of the families, the tendency to osteoporosis was due to a mutation in type I collagen. This highlights the fact that detection of mutations does not always facilitate clinical diagnosis. Moreover, manifestations not related to any syndromes may result from defects in one or more components of the extracellular matrix.
Treatment
Currently, several hormonal and pharmacological approaches to the treatment of osteogenesis imperfecta have been proposed. Prescribing calcium, calcitonin, and vitamin D supplements before obvious deficiency develops is ineffective. In young patients, in order to reduce the incidence of fractures and improve the growth of skeletal bones, the administration of bisphosphonates orally or by injection is effective. The condition of bone tissue in general cannot be improved, and there are no recommendations yet on the duration of treatment for children and the need for treatment in adults. Bone marrow transplantation to provide normal mesenchymal stem cells shows some promise. Gene therapy for autologous mesenchymal stem cells is currently being actively studied.
The article was prepared and edited by: surgeon11380 1
Osteogenesis imperfecta(osteogenesis imperfecta) - congenital fragility of bones. This complex disease of bones and some connective tissue structures, which has a wide range of changes, has been known since ancient times as a disease with a pronounced clinical picture and various forms, transmitted by inheritance. The first mentions of it appeared in the 17th century. At the end of the 18th century, i.e. 200 years ago, Olaus Jacob Ekmann described OI in members of one family, N. Ekroth (1788) reported a disease that was transmitted to children in four families, and called it osteomalacia congenita. Axmann (1831) not only described the fragility of bones in himself and his brother, but was also, apparently, the first to note such an important symptom as the presence of blue sclera.
Lobstein (1833) described bone fragility in patients of various ages. According to Vrolik (1849), fractures in children occurred either in utero or shortly after birth. E. Looser (1906) described these two forms as osteogenesis imperfecta congenita und tarda.
Many doctors studied the disease, describing more than 20 different symptoms, of which the main ones are:
changes in the structure of the skeleton and easy fractures, often small growth; blue sclera; opal-shaped dentin (dentinogenesis imperfecta); progressive deformation of the spine, chest, skull and long bones; conduction type hearing loss; hyperextension in joints and their deformation; changes in the heart and large vessels, nosebleeds, etc.
Recent work has shown that osteogenesis imperfecta is a heterogeneous hereditary disease of a genetic nature that affects connective tissue and is expressed by osteopenia and the above clinical signs.
Instead of two forms, or types, the current D.O. proposed in 1979 Sillence classification of osteogenesis imperfecta, taking into account clinical, radiological and collagen protein-gene molecular changes, is divided into 4 types.
Type I is a mild form of dominantly hereditary osteogenesis imperfecta with brittle bones and blue sclera.
Type II - perinatal-lethal.
Type III - progressive skeletal deformation.
Type IV - dominant with normal sclera and mild deformations.
P.A. Dawson et al (1999) identified type I collagen gene mutations as the cause of all four types of osteogenesis imperfecta (OI). In 2 children, radiographs showed decreased bone density in the lumbar spine and multiple fractures throughout the spine; this pathology is caused by changes in proteins, especially collagen type I. Enzymatic changes concerned the only basal mutation (1715 GA) in these children. This mutation predicts the replacement of arginine with glycine at position p43b (C43bK) in a2 (I), the father of the child had a DNA mutation of the gene. The existence of the same heterozygous mutation in 2 children suggests that the probands fully reflect this phenotype. Clinical, biochemical and molecular findings expand the understanding of the phenotype associated with type I collagen mutations causing spinal changes and dwarfism in adolescence.
Based on literary publications in recent years, as well as data presented at the 3rd International Conference on Osteogenesis Imperfecta in 1985, and the works of D.O. Sillence (1985) and others provide a brief description of these 4 types.
Type I. Osteoporosis and bone fractures are more common at an early age; after 10 years, the frequency of their occurrence decreases and increases again after 40 years. Fractures lead to bone deformation. 50% of patients experience slight growth. The blueness of the sclera is aggravated by the premature appearance of the senile rim. In some patients, the dentin is not changed, while in another part it is called opal. There are changes in the aorta and mitral heart disease, nosebleeds. In 20% of patients with OI type I, mitral valve prolapse is observed. Such a patient was described by I.A. Shamov and Sh.M. Zakharyevsky in 1989. This form is caused by structural mutations in the helical domain of pro-a, the possibility of inheritance is about 7%.
Type II. Perinatal lethal osteogenesis imperfecta. Clinically and biochemically, this is a heterogeneous group of patients, characterized by intrauterine or early neonatal death, multiplicity and ease of fractures. Divided into three groups.
Group A. The fragility of connective tissue formations is so pronounced that damage to the limbs and head of the fetus occurs even during pregnancy; the brain skull is disproportionately large, the chest is small, the limbs are shortened and curved, there are very severe degrees of calcification of the walls of the aorta and endocardium, very short height at birth (sometimes 30-25 cm).
Often premature births: in 15% of cases in the breech position, up to 20% are stillborn, the rest die either in the first days or in the 4th week of life. X-ray changes are determined in the fetus even before birth: wide femurs with wavy edges, a short chest, ribs with rosaries, etc. According to genetic data, most such cases are sporadic. Biochemical data suggest that group A patients “... are heterogeneous for mutations causing disruption of npo-oci(I) collagen chains, leading to defective triple helical assembly sequestration and incorporation into normal connective tissue. A small number of patients have heterozygous mutations in the npo-ai (I) collagen chain, while some others have been described with a single amino acid substitution, i.e. glycine to cystine, leading to the formation of disulfate bridges between the two cti(I) chains and excessive accumulation of type I collagen molecules." Examination of probands indicates a possible molecular defect that is compatible with heterozygosity of mutations in the collagen gene, which is manifested in inheritance patterns - autosomal dominant.
Group B the phenotype is similar to group A, however, respiratory system disorders are less pronounced and patients live for several years. The tubular bones are shortened and widened, the ribs are changed, but their fractures are rare. Autosomal recessive inheritance is assumed due to a recent mutation.
Group B Rarely observed, stillbirth and mortality within the first month of life are common. Patients are small in stature, the tubular bones are thin, especially the diaphysis, and there is no ossification in the bones of the brain and facial skull. Autosomal recessive inheritance is assumed.
Type III is relatively rare, the body of newborns is shortened, body weight may be normal, fractures sometimes occur during childbirth, and sometimes at the age of several years. Limb deformities (O-shaped) and kyphoscoliosis are formed, especially progressing during puberty. Changes in the skeleton and cardiovascular system lead to death in 40-50% of patients. Osteoporosis is pronounced - osteopenia, ossification and growth of bones in length are impaired, in the growth zones of bones there is uneven calcification, leading to the formation of spotting ("corn grains").
As D.O. points out. Sillence (1985), this type is characterized by autosomal recessive inheritance. In only one patient he could state that the phenotype was due to homozygosity for a molecular defect in collagen. Heredity is fresh autosomal, dominant mutation or autosomal recessive.
Type IV. Changes in the skeleton are the most common. Characterized by great variability in osteopenia, age, number of bone fractures, and blueness of the sclera (in adults, the sclera may be of normal color). The number of fractures decreases with age, normal callus formation occurs, and over the age of 30, hearing loss occurs in V3 patients. Patients with this type of osteogenesis imperfecta are divided into two groups: with sharply changed opal teeth and without changes in teeth. The predominance of autosomal dominant inheritance is expressed sharply due to the absence of a phenotypic marker (like blue sclera).
It is currently believed that osteogenesis imperfecta is caused by qualitative and quantitative changes in the synthesis of type I collagen. In type I osteogenesis imperfecta, the synthesis of structurally normal collagen is reduced, while in types II and IV, the synthesis of such collagen is normal, but due to reduced stability, the total amount of collagen is reduced. According to D.O. Sillence (1985), the number of collagen molecules produced during osteogenesis imperfecta rapidly and constantly increases, but still does not reach the norm. Therefore, he believes that in this case there is not a simple violation of collagen synthesis due to changes in the 4th chromosome, but a violation of the properties of connective tissue caused by changes in both proteoglycan synthesis and collagen gene.
D.H. Colin and R.N. Byers (1991) found that 4 patients from 60 cells synthesized a population of chain a2(I) with cystine residues in the triple helix, and clinical differences and heterogeneity in the localization of cystine residues suggest that the position and substitution sites within the chain itself are important in determining the clinical phenotype. This supports the view that patients with non-lethal osteogenesis imperfecta may often have defects in the COL A1 or COL 1A2 genes, suggesting that many of these defects are replaced by glycine residues in the oa(I) of the triple helical space.
L. Cohen-Solal et al. (1991) showed that type II and type III osteogenesis imperfecta can occur due to gonadal mosaicism. which is very important for genetic counseling in determining the appropriate phenotype of the disease.
Analyzes of type I procollagen molecules synthesized by dermal fibroblasts cultured from patients with osteogenesis imperfecta allowed us to establish two broad biochemical groups: 1) patients whose fibroblasts synthesized and effectively secreted about half the expected amount of structurally normal type I procollagen; 2) patients whose fibroblasts produced normal and abnormal populations of molecules and then secreted them.
R.J. Wenstrup et al. (1990) reported that they conducted similar studies in 224 patients and compared the biochemical data obtained with the clinical picture. It turned out that in the 1st group, where there was a decrease in the amount of normal type I procollagen, the clinical manifestations were small, and in the 2nd group, where the synthesis of normal molecules and abnormal type I procollagen was detected, the phenotype varied from moderately deforming bones to slightly a shortened figure to a disease that sharply deforms the skeleton with a moderately or sharply shortened figure. These and other studies make it possible to make a prenatal diagnosis. According to R.J. Wenstup et al. (1990), biochemical defects must be taken into account when treating.
L.M. Mikhailova (1971), during an ultramicroscopic study of bone tissue of patients with osteogenesis imperfecta, noted a reduction in the elements of granular endoplasmic reticulum in many osteoblasts, which caused a disruption of fibrillogenesis; Mitochondria also turned out to be altered, in the matrix of which there were accumulations of crystals (obviously hydroxyapatite), which, in her opinion, indicated a violation of calcium and phosphate ions. According to M.V. Volkova and N.N. Nefedyeva (1974), in patients the content of hexoses, glycoproteins, hexosamines, sialoproteins in the blood serum is sharply increased and an increased amount of mucopolysaccharides is excreted in the urine. Pathological changes in patients with osteogenesis imperfecta are very diverse.
Pseudosarcomas. After a fracture, a bone callus of large or enormous sizes develops (Fig. 5.1), sharply porotic, gradually increasing over a number of years or decades, which has to be differentiated from sarcoma, especially since in the literature there are indications of the development of osteogenic sarcoma in patients with OI . The development of pseudosarcoma is accompanied by quite severe pain, tissue tension, and local hyperemia.
The development of large callus, according to T.P. Vinogradova (1973), is a mechanism that compensates for the insufficient strength of its structures. After the fragments heal, these tumor-like calluses disappear. However, very rarely, in patients with OI, calluses do not resolve, but remain unusually large (as they were initially) or slowly continue to grow, so that they can no longer be taken as a manifestation of a compensatory process. There are no satisfactory hypotheses for their origin. We observed 3 patients with the development of “pseudosarcomas”, in 2 of which they reached gigantic sizes.
Rice. 5.1. The callus that caused the enlargement of the right femur is a pseudosarcoma.
We operated on one patient. The bone tissue had the appearance of spongiosis with thin septa and large lacunae of fatty bone marrow.
It seems that the proliferation of bone marrow leads to an increase in the volume of bone, bone lacunae, and reactive bone formation is only capable of forming thin septa and cavities, but is not able to stop the process, and therefore a normal cortical layer cannot form.
We consider it acceptable to assume that in OI the observed osteopenia is a consequence, firstly, of a slight decrease in the number of “active cells of bone tissue growth”, which, according to the theory developed by N.M. Frost et al. define bone tissue modeling; secondly, a consequence of changes in collagen structures and, thirdly, obviously, a consequence of metabolic disorders in the “third type of adipose tissue.” According to A.A. Zavarzin (1985), this type is bone marrow adipose tissue, the fat cells of which contain special lipids that are not usually used in lipid metabolism. Rapid proliferation of connective tissue, observed during fractures and the development of pseudosarcoma, contributes to the formation of large lacunae and thereby sponging of the bone: in areas where pseudosarcoma develops, sometimes the cortical layer as such is not defined.
A.N. Chernyaev and G.A. Gribanov (1982) showed that prolonged administration of calcitonin increases the synthesis by fibroblasts of not only collagen, glycosaminoglycans, but also lipids. Naturally, it is necessary to carefully study the dynamics of the level of calcitonin production in patients with pseudosarcomatous forms of osteogenesis imperfecta. We had to observe a patient with a pronounced form of pseudosarcomatous form of osteogenesis imperfecta for 30 years. It does not proceed evenly, but in stages, a period of slow calm progression is replaced by a period of rapid development, pain appears in one or another bone, the temperature rises locally, which is accompanied by the appearance of areas of hyperemia without clear boundaries, and the level of alkaline phosphatase increases sharply.
Patient A. was observed by us from the age of 33 to 61 years. She was born as a normal child in 1933 and walked independently until she was 1 year 9 months old, when her right femur was fractured. A year later, there was a second fracture of the right femur; at the age of 6 years, there was a fracture of the bones of the right tibia, then the left femur, for a total of 7 fractures. The patient was consulted by well-known specialists: G.S. Bohm, P.A. Herzen (said “he will live no more than a year”), S.M. Spasokukotsky, T.P. Krasnobaev (“this disease has no name”), I.G. Lagunova, M.K. Klimova. In 1970, she went to the CITO and was hospitalized with a diagnosis of osteogenesis imperfecta, pseudosarcomatous form.
The patient is very short (107 cm), has difficulty walking on crutches, prefers to move on a gurney. Complaints about the constantly increasing volume of the right thigh, which looked like a somewhat elongated “watermelon”, turning into the pelvis at the top and ending at the knee at the bottom. The tibia and left femur were also increased in volume. There was practically no movement in the right hip joint, and the patient could not toilet the perineum, and when urinating, urine fell on the inner surface of the thigh. We performed a subtrochanteric osteotomy of the right femur, which required not a hammer, but a chisel, which, under hand pressure, easily plunged into the bone, which represented fatty bone marrow, separated by thin bone partitions. An osteotomy of 3/4 of the diameter of the femur was performed, after which the leg was retracted outward and fixed with a plaster splint. Clinically, the pathological altered bone gave the impression of expanding bone marrow fat and osteoporotic thinning bone tissue: rare atrophic bone trabeculae.
Over the course of 25 years, there were no significant changes in the patient's condition. In 1995, a fracture of the femur occurred, after which its volume began to rapidly increase, as did the volume of the left shin; the patient had difficulty turning over in bed. Upon examination in 1997, both thighs and legs were sharply increased in volume. All the pelvic bones on both sides are enlarged, the patient’s condition is serious. A month later, they told me by phone that she had broken several ribs and was going to be admitted to the hospital. The connection was interrupted.
Treatment. Currently, it is generally accepted that for all forms of OI, treatment of osteoporosis with vitamin D3, complexones (xidifon, etc.), bisphosphonates, calcium gluconate, glycerophosphate, magnesium and potassium salts is indicated. Treatment with fish oil, vitamin D2, anabolic hormones, and ultraviolet irradiation was used less frequently [Volkov M.B., Nefedeva N.N., 1974]. The treatment developed in 1984 by N.A. was more widespread and effective. Belova in the form of a scheme and designed for 12 months (somatotropic hormone 4 units 3 times a week for the 1st and 9th months; calcitrin 3-7.5 units daily for the 2nd and 10th months; vitamin D2 - 9th and 12th months; oxidevit (vitamin D3) 1 - 1.5 mcg per day - 3rd, 4th and 11th, 12th months; festal, panzinorm, calcium gluconate. , phytin, citrate mixture, vitamins A, E, electrophoresis with calcium salts, massage, exercise therapy). According to A.P. Berezhny et al. (1988), this conservative treatment allowed us to obtain positive results: in a number of patients, fractures of long tubular bones stopped, and the treatment carried out in the preoperative period improved the results of operations. Thus, conservative treatment with vitamin D3 and other drugs should be carried out in all patients with OI.
Conservative treatment of bone fractures in this group of patients is quite a difficult task, since in some of them fractures occur frequently and sometimes are multiple. It is necessary to use all available treatment methods, and sometimes indicate indications for surgical intervention.
Given the increased fragility of bones, some orthopedists, to correct the deformity, performed osteoclasia at the apex of the curvature, corrected the deformity and fixed the limb with a plaster cast or traction.
Surgical treatment in the 40-50s was carried out in isolated patients. F.R. Bogdanov (1945) performed segmental osteotomies and used a pin he proposed for intramedullary fixation. T.S. Zatsepin used heterobone and metal pins. In 1964 M.V. Volkov proposed allogeneic grafts as an intramedullary fixation, and then developed a technique that includes decortication of deformed bone, segmental osteotomy and plastic surgery using allografts in the “bloom” type. This technique has proven to be very effective; allogeneic grafts are fused with osteogenic tissue and gradually rebuilt.
In the department we manage, surgical treatment was performed on 43 such patients, who underwent a total of 91 surgical interventions. Orthopedists involved in the surgical treatment of patients with OI have to take into account changes in the patient’s skeleton and, depending on this, set surgical goals, develop a plan and select treatment methods. We observed different clinical forms and propose to subdivide them into the following groups.
S.T.Zatsepin
Bone pathology of adults









